Building things
Operate adsorbate
The add_adsorbate function
- acat.build.action.add_adsorbate(atoms, adsorbate, site=None, surface=None, morphology=None, indices=None, height=None, composition=None, orientation=None, tilt_angle=0.0, subsurf_element=None, all_sites=None, **kwargs)[source]
A general function for adding one adsorbate to the surface. Note that this function adds one adsorbate to a random site that meets the specified condition regardless of it is already occupied or not. The function is generalized for both periodic and non-periodic systems (distinguished by atoms.pbc).
- Parameters
atoms (ase.Atoms object) – Accept any ase.Atoms object. No need to be built-in.
adsorbate (str or ase.Atom object or ase.Atoms object) – The adsorbate species to be added onto the surface.
site (str, default None) – The site type that the adsorbate should be added to.
surface (str, default None) – The surface type (crystal structure + Miller indices) If the structure is a periodic surface slab, this is required. If the structure is a nanoparticle, the function enumerates only the sites on the specified surface. For periodic slabs, a user-defined customized surface object can also be used, but note that the identified site types will only include ‘ontop’, ‘bridge’, ‘3fold’ and ‘4fold’.
morphology (str, default None) – The morphology type that the adsorbate should be added to. Only available for surface slabs.
indices (list or tuple) – The indices of the atoms that contribute to the site that you want to add adsorbate to. This has the highest priority.
height (float, default None) – The height of the added adsorbate from the surface. Use the default settings if not specified.
composition (str, default None) – The elemental of the site that should be added to.
orientation (list or numpy.array, default None) – The vector that the multidentate adsorbate is aligned to.
tilt_angle (float, default 0.) – Tilt the adsorbate with an angle (in degrees) relative to the surface normal.
subsurf_element (str, default None) – The subsurface element of the hcp or 4fold hollow site that should be added to.
all_sites (list of dicts, default None) – The list of all sites. Provide this to make the function much faster. Useful when the function is called many times.
Example 1
To add a NO molecule to a bridge site consists of one Pt and one Ni on the fcc111 surface of a bimetallic truncated octahedron:
>>> from acat.build import add_adsorbate >>> from ase.cluster import Octahedron >>> from ase.visualize import view >>> atoms = Octahedron('Ni', length=7, cutoff=2) >>> for atom in atoms: ... if atom.index % 2 == 0: ... atom.symbol = 'Pt' >>> add_adsorbate(atoms, adsorbate='NO', site='bridge', ... surface='fcc111', composition='NiPt', ... surrogate_metal='Ni') >>> view(atoms)Output:

Example 2
To add a N atom to a Ti-Ti bridge site on an anatase TiO2(101) surface. The code automatically identifies the sites occupied by surface oxygens and then adds the adsorbate to an unoccupied bridge site:
>>> from acat.build import add_adsorbate >>> from acat.settings import CustomSurface >>> from ase.spacegroup import crystal >>> from ase.build import surface >>> from ase.visualize import view >>> a, c = 3.862, 9.551 >>> anatase = crystal(['Ti', 'O'], basis=[(0 ,0, 0), (0, 0, 0.208)], ... spacegroup=141, cellpar=[a, a, c, 90, 90, 90]) >>> anatase_101_atoms = surface(anatase, (1, 0, 1), 4, vacuum=5) >>> anatase_101 = CustomSurface(anatase_101_atoms, n_layers=4) >>> # Suppose we have a surface structure resulting from some >>> # perturbation of the reference TiO2(101) surface structure >>> atoms = anatase_101_atoms.copy() >>> atoms.rattle(stdev=0.2) >>> add_adsorbate(atoms, adsorbate='N', site='bridge', ... surface=anatase_101, composition='TiTi') >>> view(atoms)Output:

The add_adsorbate_to_site function
- acat.build.action.add_adsorbate_to_site(atoms, adsorbate, site, height=None, orientation=None, tilt_angle=0.0)[source]
The base function for adding one adsorbate to a site. Site must include information of ‘normal’ and ‘position’. Useful for adding adsorbate to multiple sites or adding multidentate adsorbates.
- Parameters
atoms (ase.Atoms object) – Accept any ase.Atoms object. No need to be built-in.
adsorbate (str or ase.Atom object or ase.Atoms object) – The adsorbate species to be added onto the surface.
site (dict) – The site that the adsorbate should be added to. Must contain information of the position and the normal vector of the site.
height (float, default None) – The height of the added adsorbate from the surface. Use the default settings if not specified.
orientation (list or numpy.array, default None) – The vector that the multidentate adsorbate is aligned to.
tilt_angle (float, default None) – Tilt the adsorbate with an angle (in degrees) relative to the surface normal.
Example 1
To add CO to all fcc sites of an icosahedral nanoparticle:
>>> from acat.adsorption_sites import ClusterAdsorptionSites >>> from acat.build import add_adsorbate_to_site >>> from ase.cluster import Icosahedron >>> from ase.visualize import view >>> atoms = Icosahedron('Pt', noshells=5) >>> atoms.center(vacuum=5.) >>> cas = ClusterAdsorptionSites(atoms) >>> fcc_sites = cas.get_sites(site='fcc') >>> for site in fcc_sites: ... add_adsorbate_to_site(atoms, adsorbate='CO', site=site) >>> view(atoms)Output:

Example 2
To add a bidentate CH3OH to the (54, 57, 58) site on a Pt fcc111 surface slab and rotate to the orientation of a neighbor site:
>>> from acat.adsorption_sites import SlabAdsorptionSites >>> from acat.adsorption_sites import get_adsorption_site >>> from acat.build import add_adsorbate_to_site >>> from acat.utilities import get_mic >>> from ase.build import fcc111 >>> from ase.visualize import view >>> atoms = fcc111('Pt', (4, 4, 4), vacuum=5.) >>> i, site = get_adsorption_site(atoms, indices=(54, 57, 58), ... surface='fcc111', ... return_index=True) >>> sas = SlabAdsorptionSites(atoms, surface='fcc111') >>> sites = sas.get_sites() >>> nbsites = sas.get_neighbor_site_list(neighbor_number=1) >>> nbsite = sites[nbsites[i][0]] # Choose the first neighbor site >>> ori = get_mic(site['position'], nbsite['position'], atoms.cell) >>> add_adsorbate_to_site(atoms, adsorbate='CH3OH', site=site, ... orientation=ori) >>> view(atoms)Output:
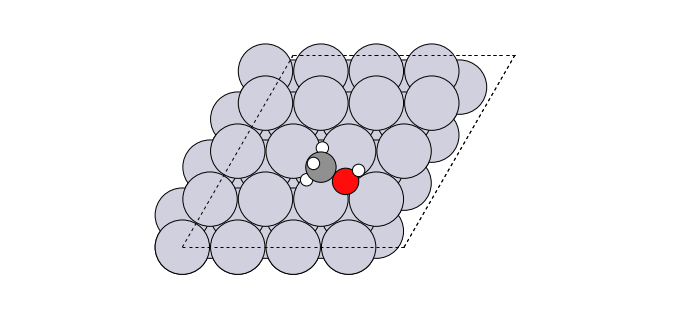
Example 3
We can also generate symmetric surface slabs with adsorbates by setting both_sides=True. Below is a more advanced example for generating a symmetric 5-layer rutile TiO2(110) surface slab with N adsorbed at all cus sites:
>>> from acat.adsorbate_coverage import SlabAdsorbateCoverage >>> from acat.build import add_adsorbate_to_site >>> from acat.settings import CustomSurface >>> from ase.spacegroup import crystal >>> from ase.build import surface >>> from ase.visualize import view >>> from collections import Counter >>> a, c = 4.584, 2.953 >>> rutile = crystal(['Ti', 'O'], basis=[(0, 0, 0), (0.3, 0.3, 0)], ... spacegroup=136, cellpar=[a, a, c, 90, 90, 90]) >>> rutile_110_atoms = surface(rutile, (1,1,0), layers=5) >>> # Remove the oxygens above the top surface >>> indices = [a.index for a in rutile_110_atoms if a.position[2] < 14.] >>> rutile_110_atoms = rutile_110_atoms[indices] >>> rutile_110_atoms.center(vacuum=5., axis=2) >>> rutile_110_atoms *= (2,3,1) >>> rutile_110 = CustomSurface(rutile_110_atoms, n_layers=5) >>> sac = SlabAdsorbateCoverage(rutile_110_atoms, surface=rutile_110, both_sides=True) >>> # Get all ontop sites which half are cus sites >>> ontop_sites = sac.get_sites(site='ontop') >>> # Get all 3fold sites occupied by oxygen >>> occupied_3fold_sites = sac.get_sites(occupied=True, site='3fold') >>> # Cus sites are then the atoms that contribute 4 times to these sites >>> occurences = [i for st in occupied_3fold_sites for i in st['indices']] >>> cus_indices = [(i,) for i, count in Counter(occurences).items() if count == 4] >>> for site in ontop_sites: ... if site['indices'] in cus_indices: ... add_adsorbate_to_site(rutile_110_atoms, adsorbate='N', site=site) >>> view(rutile_110_atoms)Output:

The add_adsorbate_to_label function
- acat.build.action.add_adsorbate_to_label(atoms, adsorbate, label, surface=None, height=None, orientation=None, tilt_angle=0.0, composition_effect=False, all_sites=None, **kwargs)[source]
Same as add_adsorbate function, except that the site type is represented by a numerical label. The function is generalized for both periodic and non-periodic systems (distinguished by atoms.pbc).
- Parameters
atoms (ase.Atoms object) – Accept any ase.Atoms object. No need to be built-in.
adsorbate (str or ase.Atom object or ase.Atoms object) – The adsorbate species to be added onto the surface.
label (int or str) – The label of the site that the adsorbate should be added to.
surface (str, default None) – The surface type (crystal structure + Miller indices) If the structure is a periodic surface slab, this is required. If the structure is a nanoparticle, the function enumerates only the sites on the specified surface. For periodic slabs, a user-defined customized surface object can also be used, but note that the identified site types will only include ‘ontop’, ‘bridge’, ‘3fold’ and ‘4fold’.
height (float, default None) – The height of the added adsorbate from the surface. Use the default settings if not specified.
orientation (list or numpy.array, default None) – The vector that the multidentate adsorbate is aligned to.
tilt_angle (float, default 0.) – Tilt the adsorbate with an angle (in degrees) relative to the surface normal.
composition_effect (bool, default False) – Whether the label is defined in bimetallic labels or not.
all_sites (list of dicts, default None) – The list of all sites. Provide this to make the function much faster. Useful when the function is called many times.
Example
To add a NH molecule to a site with bimetallic label 14 (an hcp CuCuAu site) on a bimetallic fcc110 surface slab:
>>> from acat.build import add_adsorbate_to_label >>> from ase.build import fcc110 >>> from ase.visualize import view >>> atoms = fcc110('Cu', (3, 3, 8), vacuum=5.) >>> for atom in atoms: ... if atom.index % 2 == 0: ... atom.symbol = 'Au' ... atoms.center(axis=2) >>> add_adsorbate_to_label(atoms, adsorbate='NH', ... label=14, surface='fcc110', ... composition_effect=True, ... surrogate_metal='Cu') >>> view(atoms)Output:

The remove_adsorbate_from_site function
- acat.build.action.remove_adsorbate_from_site(atoms, site, remove_fragment=False)[source]
The base function for removing one adsorbate from an occupied site. The site must include information of ‘adsorbate_indices’ or ‘fragment_indices’. Note that if you want to remove adsorbates from multiple sites, call this function multiple times will return the wrong result. Please use remove_adsorbates_from_sites instead.
- Parameters
atoms (ase.Atoms object) – Accept any ase.Atoms object. No need to be built-in.
site (dict) – The site that the adsorbate should be removed from. Must contain information of the adsorbate indices.
remove_fragment (bool, default False) – Remove the fragment of a multidentate adsorbate instead of the whole adsorbate.
Example 1
To remove a CO molecule from a fcc111 surface slab with one CO and one OH:
>>> from acat.adsorption_sites import SlabAdsorptionSites >>> from acat.adsorbate_coverage import SlabAdsorbateCoverage >>> from acat.build import add_adsorbate_to_site >>> from acat.build import remove_adsorbate_from_site >>> from ase.build import fcc111 >>> from ase.visualize import view >>> atoms = fcc111('Pt', (6, 6, 4), 4, vacuum=5.) >>> atoms.center(axis=2) >>> sas = SlabAdsorptionSites(atoms, surface='fcc111') >>> sites = sas.get_sites() >>> add_adsorbate_to_site(atoms, adsorbate='OH', site=sites[0]) >>> add_adsorbate_to_site(atoms, adsorbate='CO', site=sites[-1]) >>> sac = SlabAdsorbateCoverage(atoms, sas) >>> occupied_sites = sac.get_sites(occupied=True) >>> CO_site = next((s for s in occupied_sites if s['adsorbate'] == 'CO')) >>> remove_adsorbate_from_site(atoms, site=CO_site) >>> view(atoms)
The remove_adsorbates_from_sites function
- acat.build.action.remove_adsorbates_from_sites(atoms, sites, remove_fragments=False)[source]
The base function for removing multiple adsorbates from an occupied site. The sites must include information of ‘adsorbate_indices’ or ‘fragment_indices’.
- Parameters
atoms (ase.Atoms object) – Accept any ase.Atoms object. No need to be built-in.
sites (list of dicts) – The site that the adsorbate should be removed from. Must contain information of the adsorbate indices.
remove_fragments (bool, default False) – Remove the fragment of a multidentate adsorbate instead of the whole adsorbate.
Example 1
To remove all CO species from a fcc111 surface slab covered with both CO and OH:
>>> from acat.adsorption_sites import SlabAdsorptionSites >>> from acat.adsorbate_coverage import SlabAdsorbateCoverage >>> from acat.build.adlayer import min_dist_coverage_pattern >>> from acat.build import remove_adsorbates_from_sites >>> from ase.build import fcc111 >>> from ase.visualize import view >>> slab = fcc111('Pt', (6, 6, 4), 4, vacuum=5.) >>> slab.center(axis=2) >>> atoms = min_dist_coverage_pattern(slab, adsorbate_species=['OH','CO'], ... surface='fcc111', ... min_adsorbate_distance=5.) >>> sas = SlabAdsorptionSites(atoms, surface='fcc111') >>> sac = SlabAdsorbateCoverage(atoms, sas) >>> occupied_sites = sac.get_sites(occupied=True) >>> CO_sites = [s for s in occupied_sites if s['adsorbate'] == 'CO'] >>> remove_adsorbates_from_sites(atoms, sites=CO_sites) >>> view(atoms)Output:

Example 2
To remove O species from all cus sites on a rutile TiO2(110) surface slab:
>>> from acat.adsorbate_coverage import SlabAdsorbateCoverage >>> from acat.build import remove_adsorbates_from_sites >>> from acat.settings import CustomSurface >>> from ase.spacegroup import crystal >>> from ase.build import surface >>> from ase.visualize import view >>> from collections import Counter >>> a, c = 4.584, 2.953 >>> rutile = crystal(['Ti', 'O'], basis=[(0, 0, 0), (0.3, 0.3, 0)], ... spacegroup=136, cellpar=[a, a, c, 90, 90, 90]) >>> rutile_110_atoms = surface(rutile, (1,1,0), layers=5) >>> rutile_110_atoms.center(vacuum=5., axis=2) >>> rutile_110_atoms *= (2,3,1) >>> rutile_110 = CustomSurface(rutile_110_atoms, n_layers=5) >>> sac = SlabAdsorbateCoverage(rutile_110_atoms, surface=rutile_110) >>> # Get all ontop sites which half are cus sites >>> ontop_sites = sac.get_sites(site='ontop') >>> # Get all 3fold sites occupied by oxygen >>> occupied_3fold_sites = sac.get_sites(occupied=True, site='3fold') >>> # Cus sites are then the atoms that contribute 4 times to these sites >>> occurences = [i for st in occupied_3fold_sites for i in st['indices']] >>> cus_indices = [(i,) for i, count in Counter(occurences).items() if count == 4] >>> # Remove O from all cus sites >>> cus_sites = [s for s in ontop_sites if s['indices'] in cus_indices] >>> remove_adsorbates_from_sites(rutile_110_atoms, sites=cus_sites) >>> view(rutile_110_atoms)Output:

The remove_adsorbates_too_close function
- acat.build.action.remove_adsorbates_too_close(atoms, adsorbate_coverage=None, surface=None, min_adsorbate_distance=0.5)[source]
Find adsorbates that are too close, remove one set of them. The function is intended to remove atoms that are unphysically close. Please do not use a min_adsorbate_distace larger than 2. The function is generalized for both periodic and non-periodic systems (distinguished by atoms.pbc).
- Parameters
atoms (ase.Atoms object) – The nanoparticle or surface slab onto which the adsorbates are added. Accept any ase.Atoms object. No need to be built-in.
adsorbate_coverage (acat.adsorbate_coverage.ClusterAdsorbateCoverage object or acat.adsorbate_coverage.SlabAdsorbateCoverage object, default None) – The built-in adsorbate coverage class.
surface (str, default None) – The surface type (crystal structure + Miller indices). If the structure is a periodic surface slab, this is required. If the structure is a nanoparticle, the function only remove adsorbates from the sites on the specified surface. For periodic slabs, a user-defined customized surface object can also be used, but note that the identified site types will only include ‘ontop’, ‘bridge’, ‘3fold’ and ‘4fold’.
min_adsorbate_distance (float, default 0.) – The minimum distance between two atoms that is not considered to be to close. This distance has to be small.
Example
To remove unphysically close adsorbates on the edges of a Marks decahedron with 0.75 ML ordered CO coverage:
>>> from acat.build.adlayer import special_coverage_pattern >>> from acat.build import remove_adsorbates_too_close >>> from ase.cluster import Decahedron >>> from ase.visualize import view >>> atoms = Decahedron('Pt', p=4, q=3, r=1) >>> atoms.center(vacuum=5.) >>> pattern = special_coverage_pattern(atoms, adsorbate='CO', ... coverage=0.75) >>> remove_adsorbates_too_close(pattern, min_adsorbate_distance=1.) >>> view(pattern)Output:

Generate adsorbate overlayer patterns
The RandomPatternGenerator class
- class acat.build.adlayer.RandomPatternGenerator(images, adsorbate_species, image_probabilities=None, species_probabilities=None, min_adsorbate_distance=1.5, adsorption_sites=None, heights={'3fold': 1.3, '4fold': 1.3, '5fold': 1.5, '6fold': 0.0, 'bridge': 1.5, 'fcc': 1.3, 'hcp': 1.3, 'longbridge': 1.5, 'ontop': 1.8, 'shortbridge': 1.5}, subtract_heights=False, dmax=2.5, species_forbidden_sites=None, species_forbidden_labels=None, fragmentation=True, trajectory='patterns.traj', append_trajectory=False, logfile='patterns.log', **kwargs)[source]
RandomPatternGenerator is a class for generating adsorbate overlayer patterns stochastically. Graph automorphism is implemented to identify identical adlayer patterns. 4 adsorbate actions are supported: add, remove, move, replace. The class is generalized for both periodic and non-periodic systems (distinguished by atoms.pbc).
- Parameters
images (ase.Atoms object or list of ase.Atoms objects) – The structure to perform the adsorbate actions on. If a list of structures is provided, perform one adsorbate action on one of the structures in each step. Accept any ase.Atoms object. No need to be built-in.
adsorbate_species (str or list of strs) – A list of adsorbate species to be randomly added to the surface.
image_probabilities (listt, default None) – A list of the probabilities of selecting each structure. Selecting structure with equal probability if not specified.
species_probabilities (dict, default None) – A dictionary that contains keys of each adsorbate species and values of their probabilities of adding onto the surface. Adding adsorbate species with equal probability if not specified.
min_adsorbate_distance (float, default 1.5) – The minimum distance constraint between two atoms that belongs to two adsorbates.
adsorption_sites (acat.adsorption_sites.ClusterAdsorptionSites object or acat.adsorption_sites.SlabAdsorptionSites object or the corresponding pickle file path, default None) – Provide the built-in adsorption sites class to accelerate the pattern generation. Make sure all the structures have the same periodicity and atom indexing. If composition_effect=True, you should only provide adsorption_sites when the surface composition is fixed. If this is not provided, the arguments for identifying adsorption sites can still be passed in by **kwargs.
heights (dict, default acat.settings.site_heights) – A dictionary that contains the adsorbate height for each site type. Use the default height settings if the height for a site type is not specified.
subtract_heights (bool, default False) – Whether to subtract the height from the bond length when allocating a site to an adsorbate. Default is to allocate the site that is closest to the adsorbate’s binding atom without subtracting height. Useful for ensuring the allocated site for each adsorbate is consistent with the site to which the adsorbate was added.
dmax (float, default 2.5) – The maximum bond length (in Angstrom) between an atom and its nearest site to be considered as the atom being bound to the site.
species_forbidden_sites (dict, default None) – A dictionary that contains keys of each adsorbate species and values of the site (can be one or multiple site types) that the speices is not allowed to add to. All sites are availabe for a species if not specified. Note that this does not differentiate sites with different compositions.
species_forbidden_labels (dict, default None) – Same as species_forbidden_sites except that the adsorption sites are written as numerical labels according to acat.labels. Useful when you need to differentiate sites with different compositions.
fragmentation (bool, default True) – Whether to cut multidentate species into fragments. This ensures that multidentate species with different orientations are considered as different adlayer patterns.
trajectory (str, default 'patterns.traj') – The name of the output ase trajectory file.
append_trajectory (bool, default False) –
Whether to append structures to the existing trajectory. If only unique patterns are accepted, the code will also check graph automorphism for the existing structures in the trajectory. This is also useful when you want to generate adlayer patterns stochastically but for all images systematically, e.g. generating 10 stochastic adlayer patterns for each image:
>>> from acat.build.adlayer import RandomPatternGenerator as RPG >>> for atoms in images: ... rpg = RPG(atoms, ..., append_trajectory=True) ... rpg.run(num_gen = 10)logfile (str, default 'patterns.log') – The name of the log file.
- run(num_gen, action='add', action_probabilities=None, num_act=1, add_species_composition=None, unique=False, hmax=2, site_preference=None, subsurf_effect=False)[source]
Run the pattern generator.
- Parameters
num_gen (int) – Number of patterns to generate.
action (str or list of strs, default 'add') – Action(s) to perform. If a list of actions is provided, select actions from the list randomly or with probabilities.
action_probabilities (dict, default None) – A dictionary that contains keys of each action and values of the corresponding probabilities. Select actions with equal probability if not specified.
num_act (int, default 1) – Number of times performed for each action. Useful for operating more than one adsorbates at a time. This becomes extremely slow when adding many adsorbates to generate high coverage patterns. The recommended ways to generate high coverage patterns are: 1) adding one adsorbate at a time from low to high coverage if you want to control the exact number of adsorbates; 2) use acat.build.adlayer.min_dist_coverage_pattern if you want to control the minimum adsorbate distance. This is the fastest way, but the number of adsorbates is not guaranteed to be fixed.
add_species_composition (dict, default None) – A dictionary that contains keys of each adsorbate species and values of the species composition to be added onto the surface. Adding adsorbate species according to species_probabilities if not specified. Please only use this if the action is ‘add’.
unique (bool, default False) – Whether to discard duplicate patterns based on graph automorphism. The Weisfeiler-Lehman subtree kernel is used to check identity.
hmax (int, default 2) – Maximum number of iterations for color refinement. Only relevant if unique=True.
site_preference (str or list of strs, defualt None) – The site type(s) that has higher priority to attach adsorbates.
subsurf_effect (bool, default False) – Whether to take subsurface atoms into consideration when checking uniqueness. Could be important for surfaces like fcc100.
Example 1
The following example illustrates how to stochastically generate 100 unique adsorbate overlayer patterns with one more adsorbate chosen from CO, OH, CH3 and CHO, based on 10 Pt fcc111 surface slabs with random C and O coverages, where CH3 is forbidden to be added to ontop and bridge sites:
>>> from acat.build.adlayer import RandomPatternGenerator as RPG >>> from acat.build.adlayer import min_dist_coverage_pattern >>> from ase.build import fcc111 >>> from ase.io import read >>> from ase.visualize import view >>> slab = fcc111('Pt', (6, 6, 4), 4, vacuum=5.) >>> slab.center(axis=2) >>> images = [] >>> for _ in range(10): ... atoms = slab.copy() ... image = min_dist_coverage_pattern(atoms, adsorbate_species=['C','O'], ... surface='fcc111', ... min_adsorbate_distance=5.) ... images.append(image) >>> rpg = RPG(images, adsorbate_species=['CO','OH','CH3','CHO'], ... species_probabilities={'CO': 0.3, 'OH': 0.3, ... 'CH3': 0.2, 'CHO': 0.2}, ... min_adsorbate_distance=1.5, ... surface='fcc111', ... composition_effect=False, ... species_forbidden_sites={'CH3': ['ontop','bridge']}) >>> rpg.run(num_gen=100, action='add', unique=True) >>> images = read('patterns.traj', index=':') >>> view(images)Output:

Example 2
The following example illustrates how to generate 20 unique coverage patterns, each adding 4 adsorbates (randomly chosen from H, OH and H2O) onto a fcc100 Ni2Cu surface slab on both top and bottom interfaces (
bulk waterin between) with probabilities of 0.25, 0.25, 0.5, respectively, and a minimum adsorbate distance of 2.5 Angstrom:>>> from acat.build.adlayer import RandomPatternGenerator as RPG >>> from acat.adsorption_sites import SlabAdsorptionSites >>> from ase.io import read >>> from ase.build import fcc100 >>> from ase.visualize import view >>> water_bulk = read('water_bulk.xyz') >>> water_bulk.center(vacuum=11., axis=2) >>> slab = fcc100('Ni', (4, 4, 8), vacuum=9.5, periodic=True) >>> for atom in slab: ... if atom.index % 3 == 0: ... atom.symbol = 'Cu' >>> slab.translate(-slab.cell[2] / 2) >>> slab.wrap() >>> atoms = slab + water_bulk >>> sas = SlabAdsorptionSites(atoms, surface='fcc100', ... composition_effect=True, ... both_sides=True, ... surrogate_metal='Ni') >>> rpg = RPG(atoms, adsorbate_species=['H','OH','OH2'], ... species_probabilities={'H': 0.25, 'OH': 0.25, 'OH2': 0.5}, ... min_adsorbate_distance=2.5, ... adsorption_sites=sas, ... surface='fcc100') >>> rpg.run(num_gen=20, action='add', num_act=4, unique=True) >>> images = read('patterns.traj', index=':') >>> view(images)Output:

Example 3
The following example illustrates how to generate 20 random adsorbate overlayer patterns with 5 adsorbates: 1 CO, 2 OH and 2 N, on 10 quaternary cuboctahedral nanoalloys with random chemical orderings. The minimum adsorbate distance is set to 3 Angstrom and duplicate patterns are allowed (very unlikely for nanoparticles):
>>> from acat.adsorption_sites import ClusterAdsorptionSites >>> from acat.build.adlayer import RandomPatternGenerator as RPG >>> from acat.build.ordering import RandomOrderingGenerator as ROG >>> from ase.cluster import Octahedron >>> from ase.io import read >>> from ase.visualize import view >>> atoms = Octahedron('Ni', length=5, cutoff=2) >>> atoms.center(vacuum=5.) >>> rog = ROG(atoms, elements=['Ni', 'Cu', 'Pt', 'Au']) >>> rog.run(num_gen=10) >>> particles = read('orderings.traj', index=':') >>> rpg = RPG(particles, adsorbate_species=['CO','OH','N'], ... min_adsorbate_distance=3., ... composition_effect=True, ... surrogate_metal='Ni') >>> rpg.run(num_gen=20, action='add', num_act=5, unique=False, ... add_species_composition={'CO': 1, 'OH': 2, 'N': 2}) >>> images = read('patterns.traj', index=':') >>> view(images)Output:

The SystematicPatternGenerator class
- class acat.build.adlayer.SystematicPatternGenerator(images, adsorbate_species, min_adsorbate_distance=1.5, adsorption_sites=None, heights={'3fold': 1.3, '4fold': 1.3, '5fold': 1.5, '6fold': 0.0, 'bridge': 1.5, 'fcc': 1.3, 'hcp': 1.3, 'longbridge': 1.5, 'ontop': 1.8, 'shortbridge': 1.5}, subtract_heights=False, dmax=2.5, species_forbidden_sites=None, species_forbidden_labels=None, smart_skip=True, enumerate_orientations=True, trajectory='patterns.traj', append_trajectory=False, logfile='patterns.log', **kwargs)[source]
SystematicPatternGenerator is a class for generating adsorbate overlayer patterns systematically. This is useful to enumerate all unique patterns at low coverage, but explodes at higher coverages. Graph automorphism is implemented to identify identical adlayer patterns. 4 adsorbate actions are supported: add, remove, move, replace. The class is generalized for both periodic and non-periodic systems (distinguished by atoms.pbc).
- Parameters
images (ase.Atoms object or list of ase.Atoms objects) – The structure to perform the adsorbate actions on. If a list of structures is provided, perform one adsorbate action on one of the structures in each step. Accept any ase.Atoms object. No need to be built-in.
adsorbate_species (str or list of strs) – A list of adsorbate species to be randomly added to the surface.
min_adsorbate_distance (float, default 1.5) – The minimum distance constraint between two atoms that belongs to two adsorbates.
adsorption_sites (acat.adsorption_sites.ClusterAdsorptionSites object or acat.adsorption_sites.SlabAdsorptionSites object or the corresponding pickle file path, default None) – Provide the built-in adsorption sites class to accelerate the pattern generation. Make sure all the structures have the same periodicity and atom indexing. If composition_effect=True, you should only provide adsorption_sites when the surface composition is fixed. If this is not provided, the arguments for identifying adsorption sites can still be passed in by **kwargs.
heights (dict, default acat.settings.site_heights) – A dictionary that contains the adsorbate height for each site type. Use the default height settings if the height for a site type is not specified.
subtract_heights (bool, default False) – Whether to subtract the height from the bond length when allocating a site to an adsorbate. Default is to allocate the site that is closest to the adsorbate’s binding atom without subtracting height. Useful for ensuring the allocated site for each adsorbate is consistent with the site to which the adsorbate was added.
dmax (float, default 2.5) – The maximum bond length (in Angstrom) between an atom and its nearest site to be considered as the atom being bound to the site.
species_forbidden_sites (dict, default None) – A dictionary that contains keys of each adsorbate species and values of the site (can be one or multiple site types) that the speices is not allowed to add to. All sites are availabe for a species if not specified. Note that this does not differentiate sites with different compositions.
species_forbidden_labels (dict, default None) – Same as species_forbidden_sites except that the adsorption sites are written as numerical labels according to acat.labels. Useful when you need to differentiate sites with different compositions.
smart_skip (bool, default True) – Whether to smartly skipping sites in the neighboring shell when populating a site with an adsorabte. This could potentially speed up the enumeration by a lot. Note that this should be set to False for metal oxide surfaces.
enumerate_orientations (bool, default True) – Whether to enumerate all orientations of multidentate species. This ensures that multidentate species with different orientations are all enumerated.
trajectory (str, default 'patterns.traj') – The name of the output ase trajectory file.
append_trajectory (bool, default False) – Whether to append structures to the existing trajectory. If only unique patterns are accepted, the code will also check graph automorphism for the existing structures in the trajectory.
logfile (str, default 'patterns.log') – The name of the log file.
- run(max_gen_per_image=None, action='add', num_act=1, add_species_composition=None, unique=False, hmax=2, site_preference=None, subsurf_effect=False)[source]
Run the pattern generator.
- Parameters
max_gen_per_image (int, default None) – Maximum number of patterns to generate for each image. Enumerate all possible patterns if not specified.
action (str, defualt 'add') – Action to perform.
num_act (int, default 1) – Number of times performed for the action. Useful for operating more than one adsorbates at a time. This becomes extremely slow when adding many adsorbates to generate high coverage patterns. The recommended way to generate high coverage patterns is to add one adsorbate at a time from low to high coverage if you want to control the exact number of adsorbates.
add_species_composition (dict, default None) – A dictionary that contains keys of each adsorbate species and values of the species composition to be added onto the surface. Adding all possible adsorbate species if not specified. Please only use this if the action is ‘add’.
unique (bool, default False) – Whether to discard duplicate patterns based on graph automorphism. The Weisfeiler-Lehman subtree kernel is used to check identity.
hmax (int, default 2) – Maximum number of iterations for color refinement. Only relevant if unique=True.
site_preference (str or list of strs, defualt None) – The site type(s) that has higher priority to attach adsorbates.
subsurf_effect (bool, default False) – Whether to take subsurface atoms into consideration when checking uniqueness. Could be important for surfaces like fcc100.
Example 1
The following example illustrates how to add CO to all unique sites on a cuboctahedral bimetallic nanoparticle with a minimum adsorbate distance of 2 Angstrom:
>>> from acat.adsorption_sites import ClusterAdsorptionSites >>> from acat.build.adlayer import SystematicPatternGenerator as SPG >>> from ase.cluster import Octahedron >>> from ase.io import read >>> from ase.visualize import view >>> atoms = Octahedron('Cu', length=7, cutoff=3) >>> for atom in atoms: ... if atom.index % 2 == 0: ... atom.symbol = 'Au' >>> atoms.center(vacuum=5.) >>> cas = ClusterAdsorptionSites(atoms, composition_effect=True, ... surrogate_metal='Cu') >>> spg = SPG(atoms, adsorbate_species='CO', ... min_adsorbate_distance=2., ... adsorption_sites=cas, ... composition_effect=True) >>> spg.run(action='add') >>> images = read('patterns.traj', index=':') >>> view(images)Output:

Example 2
The following example illustrates how to enumerate all unique coverage patterns consists of 3 adsorbates: 1 C, 1 N and 1 O on a bimetallic bcc111 surface slab with a minimum adsorbate distance of 2 Angstrom (here only generate a maximum of 100 unique patterns):
>>> from acat.build.adlayer import SystematicPatternGenerator as SPG >>> from acat.adsorption_sites import SlabAdsorptionSites >>> from ase.io import read >>> from ase.build import bcc111 >>> from ase.visualize import view >>> atoms = bcc111('Fe', (2, 2, 12), vacuum=5.) >>> for atom in atoms: ... if atom.index % 2 == 0: ... atom.symbol = 'Mo' >>> atoms.center(axis=2) >>> sas = SlabAdsorptionSites(atoms, surface='bcc111', ... ignore_sites='3fold', ... composition_effect=True) >>> spg = SPG(atoms, adsorbate_species=['C','N','O'], ... min_adsorbate_distance=2., ... adsorption_sites=sas, ... surface='bcc111', ... composition_effect=True) >>> spg.run(max_gen_per_image=100, action='add', num_act=3, ... add_species_composition={'C': 1, 'N': 1, 'O': 1}) >>> images = read('patterns.traj', index=':') >>> view(images)Output:
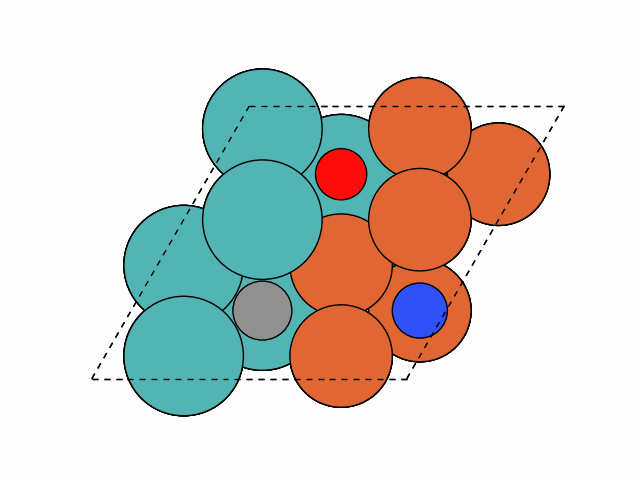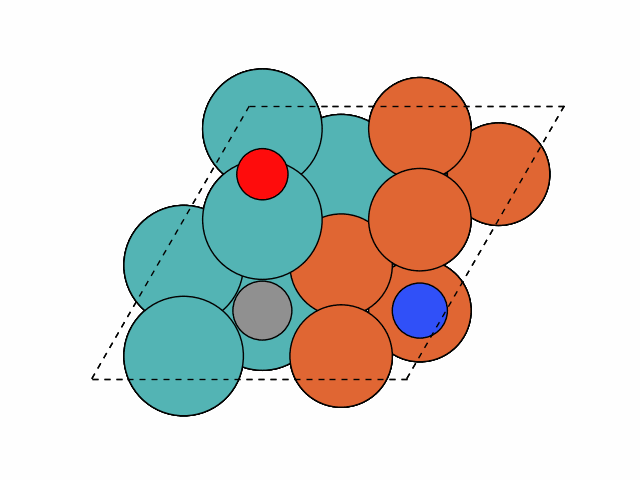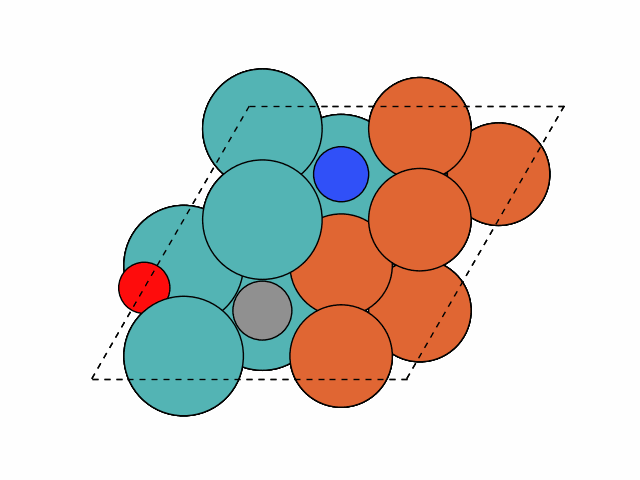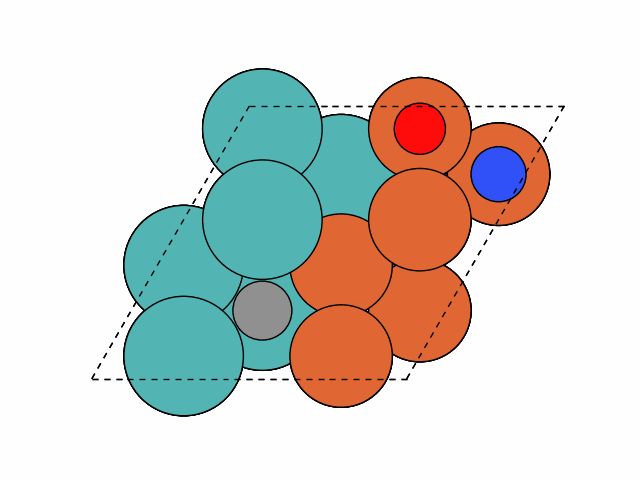
Example 3
The following example illustrates how to enumerate all unique sites on a (
Cu2O(111) surface slab) that considers the lattice-O effect on the local environment of each site. Here we will also enumerate all unique orientations of a multidentate SO2 adsorbate at these unique sites:>>> from acat.build.adlayer import SystematicPatternGenerator as SPG >>> from acat.adsorption_sites import SlabAdsorptionSites >>> from acat.settings import CustomSurface >>> from ase.io import read >>> from ase.visualize import view >>> atoms = read('Cu2O_111.xyz') >>> Cu2O_111 = CustomSurface(atoms, n_layers=4) >>> sas = SlabAdsorptionSites(atoms, surface=Cu2O_111) >>> spg = SPG(atoms, adsorbate_species='SO2', ... min_adsorbate_distance=1.5, ... adsorption_sites=sas, ... smart_skip=False, # Set this to False for oxides ... enumerate_orientations=True, ... trajectory='Cu2O_111_SO2_unique_configs.traj') >>> spg.run(action='add', num_act=1, unique=True) >>> images = read('Cu2O_111_SO2_unique_configs.traj', index=':') >>> view(images)Output:

The OrderedPatternGenerator class
- class acat.build.adlayer.OrderedPatternGenerator(images, adsorbate_species, species_probabilities=None, repeating_distance=None, max_species=None, sorting_vector=array([1, 0]), adsorption_sites=None, remove_site_shells=1, remove_site_radius=None, populate_isolated_sites=False, allow_odd=False, heights={'3fold': 1.3, '4fold': 1.3, '5fold': 1.5, '6fold': 0.0, 'bridge': 1.5, 'fcc': 1.3, 'hcp': 1.3, 'longbridge': 1.5, 'ontop': 1.8, 'shortbridge': 1.5}, subtract_heights=False, site_groups=None, save_groups=False, fragmentation=True, dmax=2.5, dtol=0.3, trajectory='patterns.traj', append_trajectory=False, **kwargs)[source]
OrderedPatternGenerator is a class for generating adsorbate overlayer patterns stochastically. Graph automorphism is implemented to identify identical adlayer patterns. 4 adsorbate actions are supported: add, remove, move, replace. The class is generalized for both periodic and non-periodic systems (distinguished by atoms.pbc).
- Parameters
images (ase.Atoms object or list of ase.Atoms objects) – The structure to perform the adsorbate actions on. If a list of structures is provided, perform one adsorbate action on one of the structures in each step. Accept any ase.Atoms object. No need to be built-in.
adsorbate_species (str or list of strs) – A list of possible adsorbate species to be added to the surface.
species_probabilities (dict, default None) – A dictionary that contains keys of each adsorbate species and values of their probabilities of adding onto the surface.
repeating_distance (float, default None) – The pairwise distance (in Angstrom) between two symmetry-equivalent adsorption sites. If repeating_distance is not provided, all possible repeating distances are considered.
max_species (int, default None) – The maximum allowed adsorbate species (excluding vacancies) for a single structure. Allow all adsorbatae species if not specified.
sorting_vector (numpy.array, default numpy.array([1, 0])) – The 2D (or 3D) vector [x, y] represeting the vertical plane to sort the sites based on the signed distance from the site to that plane before grouping. Use the x-axis by default. Recommend using default or the diagonal vector.
adsorption_sites (acat.adsorption_sites.ClusterAdsorptionSites object or acat.adsorption_sites.SlabAdsorptionSites object or the corresponding pickle file path, default None) – Provide the built-in adsorption sites class to accelerate the pattern generation. Make sure all the structures have the same periodicity and atom indexing. If composition_effect=True, you should only provide adsorption_sites when the surface composition is fixed. If this is not provided, the arguments for identifying adsorption sites can still be passed in by **kwargs.
remove_site_shells (int, default 1) – The neighbor shell number within which the neighbor sites should be removed. Remove the 1st neighbor site shell by default. Set to 0 if no site should be removed.
remove_site_radius (float, default None) – The radius within which the neighbor sites should be removed. This serves as an alternative to remove_site_shells.
populate_isolated_sites (bool, default False) – Whether to add adsorbates to low-symmetry sites that are not grouped with any other sites.
allow_odd (bool, default False) – Whether to allow odd number of adsorbates. This is done by singling out one site for each symmetry-inequivalent site.
heights (dict, default acat.settings.site_heights) – A dictionary that contains the adsorbate height for each site type. Use the default height settings if the height for a site type is not specified.
subtract_heights (bool, default False) – Whether to subtract the height from the bond length when allocating a site to an adsorbate. Default is to allocate the site that is closest to the adsorbate’s binding atom without subtracting height. Useful for ensuring the allocated site for each adsorbate is consistent with the site to which the adsorbate was added.
site_groups (list of lists, default None) – Provide the user defined symmetry equivalent site groups as a list of lists of site indices (of the site list). Useful for generating structures with symmetries that are not supported.
save_groups (bool, default False) – Whether to save the site groups in atoms.info[‘data’][‘groups’] for each generated structure. If there is groups present (e.g. the groups of the slab atoms), append the site groups to it.
fragmentation (bool, default True) – Whether to cut multidentate species into fragments. This ensures that multidentate species with different orientations are considered as different adlayer patterns.
dmax (float, default 2.5) – The maximum bond length (in Angstrom) between an atom and its nearest site to be considered as the atom being bound to the site.
dtol (float, default 0.3) – The tolerance (in Angstrom) when calculating the repeating distance.
trajectory (str, default 'patterns.traj') – The name of the output ase trajectory file.
append_trajectory (bool, default False) – Whether to append structures to the existing trajectory. If only unique patterns are accepted, the code will also check graph automorphism for the existing structures in the trajectory.
- get_site_groups(return_all_site_groups=False)[source]
Get the groups (a list of lists of site indices) of all pairs of symmetry-equivalent sites.
- Parameters
return_all_site_groups (bool, default False) – Whether to return all possible high-symmetry groupings of the adsorption sites.
- run(max_gen=None, unique=False, hmax=2)[source]
Run the ordered pattern generator.
- Parameters
max_gen (int, default None) – Maximum number of chemical orderings to generate. Running forever (until exhaustive for systematic search) if not specified.
unique (bool, default False) – Whether to discard duplicate patterns based on graph automorphism. The Weisfeiler-Lehman subtree kernel is used to check identity.
hmax (int, default 2) – Maximum number of iterations for color refinement. Only relevant if unique=True.
Example 1
The following example illustrates how to generate 50 unique ordered adlayer patterns on a fcc111 NiCu surface slab with possible adsorbates of C, N, O, OH with a repeating distance of 5.026 Angstrom, where each structure is limited to have at most 2 different adsorbate species, and the neighbor sites around each occupied site must be removed:
>>> from acat.build.adlayer import OrderedPatternGenerator as OPG >>> from acat.adsorption_sites import SlabAdsorptionSites >>> from ase.io import read >>> from ase.build import fcc111 >>> from ase.visualize import view >>> atoms = fcc111('Ni', (4, 4, 4), vacuum=5.) >>> for atom in atoms: ... if atom.index % 2 == 0: ... atom.symbol = 'Cu' >>> atoms.center(axis=2) >>> sas = SlabAdsorptionSites(atoms, surface='fcc111', ... allow_6fold=False, ... ignore_sites='bridge', ... surrogate_metal='Ni') >>> opg = OPG(atoms, adsorbate_species=['C', 'N', 'O', 'OH'], ... surface='fcc111', ... repeating_distance=5.026, ... max_species=2, ... adsorption_sites=sas, ... remove_site_shells=1) >>> opg.run(max_gen=50, unique=True) >>> images = read('patterns.traj', index=':') >>> view(images)Output:

Example 2
The following example illustrates how to generate 50 unique ordered adlayer patterns on a fcc100 NiCu surface slab with possible adsorbates of C, N, O, OH with a repeating distance of 5.026 Angstrom, where each structure is limited to have at most 2 different adsorbate species, the 1st and 2nd neighbor sites around each occupied site must be removed, and the sites are sorted according to the diagonal vector:
>>> from acat.build.adlayer import OrderedPatternGenerator as OPG >>> from acat.adsorption_sites import SlabAdsorptionSites >>> from ase.io import read >>> from ase.build import fcc100 >>> from ase.visualize import view >>> atoms = fcc100('Ni', (4, 4, 4), vacuum=5.) ... for atom in atoms: ... if atom.index % 2 == 0: ... atom.symbol = 'Cu' >>> atoms.center(axis=2) >>> diagonal_vec = atoms[63].position - atoms[48].position >>> sas = SlabAdsorptionSites(atoms, surface='fcc100', ... allow_6fold=False, ... ignore_sites=None, ... surrogate_metal='Ni') >>> opg = OPG(atoms, adsorbate_species=['C', 'N', 'O', 'OH'], ... surface='fcc100', ... repeating_distance=5.026, ... max_species=2, ... sorting_vector=diagonal_vec, ... adsorption_sites=sas, ... remove_site_shells=2) >>> opg.run(max_gen=50, unique=True) >>> images = read('patterns.traj', index=':') >>> view(images)Output:

The special_coverage_pattern function
- acat.build.adlayer.special_coverage_pattern(atoms, adsorbate_species, coverage=1.0, species_probabilities=None, adsorption_sites=None, surface=None, height=None, min_adsorbate_distance=0.0, **kwargs)[source]
A function for generating representative ordered adsorbate overlayer patterns. The function is generalized for both periodic and non-periodic systems (distinguished by atoms.pbc). Currently only clean metal surfaces/nanonparticles are supported.
- Parameters
atoms (ase.Atoms object) – The nanoparticle or surface slab onto which the adsorbates are added. Accept any ase.Atoms object. No need to be built-in.
adsorbate_species (str or list of strs) – A list of adsorbate species to be added to the surface.
coverage (float, default 1.) – The coverage (ML) of the adsorbate (N_adsorbate / N_surf_atoms). Support 4 adlayer patterns ( 0.25 for p(2x2) pattern; 0.5 for c(2x2) pattern on fcc100 or honeycomb pattern on fcc111; 0.75 for (2x2) pattern on fcc100 or Kagome pattern on fcc111; 1. for p(1x1) pattern; 2. for ontop+4fold pattern on fcc100 or fcc+hcp pattern on fcc111. Note that for small nanoparticles, the function might give results that do not correspond to the coverage. This is normal since the surface area can be too small to encompass the adlayer pattern properly. We expect this function to work well on large nanoparticles and surface slabs.
species_probabilities (dict, default None) – A dictionary that contains keys of each adsorbate species and values of their probabilities of adding onto the surface.
adsorption_sites (acat.adsorption_sites.ClusterAdsorptionSites object or acat.adsorption_sites.SlabAdsorptionSites object or the corresponding pickle file path, default None) – Provide the built-in adsorption sites class to accelerate the pattern generation. If this is not provided, the arguments for identifying adsorption sites can still be passed in by **kwargs.
surface (str, default None) – The surface type (crystal structure + Miller indices). For now only support 2 common surfaces: fcc100 and fcc111. If the structure is a periodic surface slab, this is required when adsorption_sites is not provided. If the structure is a nanoparticle, the function only add adsorbates to the sites on the specified surface. For periodic slabs, a user-defined customized surface object can also be used, but note that the identified site types will only include ‘ontop’, ‘bridge’, ‘3fold’ and ‘4fold’.
height (float, default None) – The height of the added adsorbate from the surface. Use the default settings if not specified.
min_adsorbate_distance (float, default 0.) – The minimum distance between two atoms that belongs to two adsorbates.
Example 1
To add a 0.5 ML CO adlayer pattern on a cuboctahedron:
>>> from acat.build.adlayer import special_coverage_pattern as scp >>> from ase.cluster import Octahedron >>> from ase.visualize import view >>> atoms = Octahedron('Au', length=9, cutoff=4) >>> atoms.center(vacuum=5.) >>> pattern = scp(atoms, adsorbate_species='CO', coverage=0.5) >>> view(pattern)Output:

Example 2
To add a 0.5 ML CO adlayer pattern on a fcc111 surface slab:
>>> from acat.build.adlayer import special_coverage_pattern as scp >>> from ase.build import fcc111 >>> from ase.visualize import view >>> atoms = fcc111('Cu', (8, 8, 4), vacuum=5.) >>> atoms.center(axis=2) >>> pattern = scp(atoms, adsorbate_species='CO', ... coverage=0.5, surface='fcc111') >>> view(pattern)Output:

The max_dist_coverage_pattern function
- acat.build.adlayer.max_dist_coverage_pattern(atoms, adsorbate_species, coverage, site_types=None, species_probabilities=None, adsorption_sites=None, surface=None, heights={'3fold': 1.3, '4fold': 1.3, '5fold': 1.5, '6fold': 0.0, 'bridge': 1.5, 'fcc': 1.3, 'hcp': 1.3, 'longbridge': 1.5, 'ontop': 1.8, 'shortbridge': 1.5}, **kwargs)[source]
A function for generating random adlayer patterns with a certain surface adsorbate coverage (i.e. fixed number of adsorbates N) and trying to even the adsorbate density. The function samples N sites from all given sites using K-medoids clustering to maximize the minimum distance between sites and add N adsorbates to these sites. The function is generalized for both periodic and non-periodic systems (distinguished by atoms.pbc). pyclustering is required. Currently only clean metal surfaces/nanoparticles are supported.
- Parameters
atoms (ase.Atoms object) – The nanoparticle or surface slab onto which the adsorbates are added. Accept any ase.Atoms object. No need to be built-in.
adsorbate_species (str or list of strs) – A list of adsorbate species to be added to the surface.
coverage (float) – The surface coverage calculated by (number of adsorbates / number of surface atoms). Subsurface sites are not considered.
site_types (str or list of strs, default None) – The site type(s) that the adsorbates should be added to. Consider all sites if not specified.
species_probabilities (dict, default None) – A dictionary that contains keys of each adsorbate species and values of their probabilities of adding onto the surface.
adsorption_sites (acat.adsorption_sites.ClusterAdsorptionSites object or acat.adsorption_sites.SlabAdsorptionSites object or the corresponding pickle file path, default None) – Provide the built-in adsorption sites class to accelerate the pattern generation. If this is not provided, the arguments for identifying adsorption sites can still be passed in by **kwargs.
surface (str, default None) – The surface type (crystal structure + Miller indices). If the structure is a periodic surface slab, this is required when adsorption_sites is not provided. If the structure is a nanoparticle, the function only add adsorbates to the sites on the specified surface. For periodic slabs, a user-defined customized surface object can also be used, but note that the identified site types will only include ‘ontop’, ‘bridge’, ‘3fold’ and ‘4fold’.
heights (dict, default acat.settings.site_heights) – A dictionary that contains the adsorbate height for each site type. Use the default height settings if the height for a site type is not specified.
Example 1
To add 0.33 ML CO to all fcc and hcp sites on an icosahedron:
>>> from acat.build.adlayer import max_dist_coverage_pattern as maxdcp >>> from ase.cluster import Icosahedron >>> from ase.visualize import view >>> atoms = Icosahedron('Au', noshells=5) >>> atoms.center(vacuum=5.) >>> pattern = maxdcp(atoms, adsorbate_species='CO', ... coverage=0.33, site_types=['fcc','hcp']) >>> view(pattern)Output:
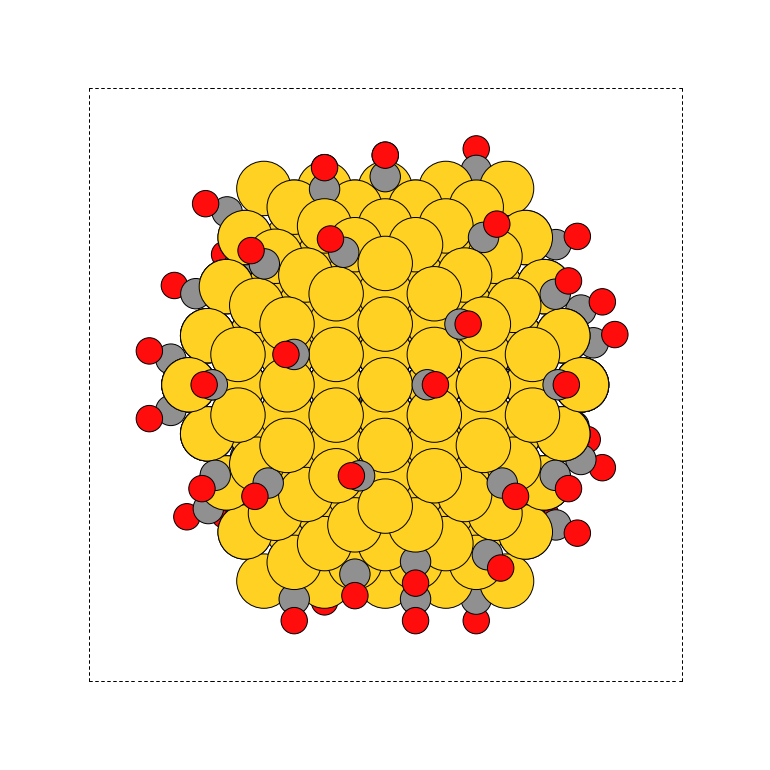
Example 2
To add N and O (3 : 1 ratio) to all 3fold sites on a bcc110 surface slab:
>>> from acat.build.adlayer import max_dist_coverage_pattern as maxdcp >>> from ase.build import bcc110 >>> from ase.visualize import view >>> atoms = bcc110('Mo', (8, 8, 4), vacuum=5.) >>> atoms.center(axis=2) >>> pattern = maxdcp(atoms, adsorbate_species=['N','O'], ... species_probabilities={'N': 0.75, 'O':0.25}, ... coverage=1, site_types='3fold', surface='bcc110') >>> view(pattern)Output:

The min_dist_coverage_pattern function
- acat.build.adlayer.min_dist_coverage_pattern(atoms, adsorbate_species, species_probabilities=None, adsorption_sites=None, surface=None, min_adsorbate_distance=1.5, site_types=None, heights={'3fold': 1.3, '4fold': 1.3, '5fold': 1.5, '6fold': 0.0, 'bridge': 1.5, 'fcc': 1.3, 'hcp': 1.3, 'longbridge': 1.5, 'ontop': 1.8, 'shortbridge': 1.5}, site_preference=None, **kwargs)[source]
A function for generating random adlayer patterns with a minimum distance constraint and trying to maximize the adsorbate density. The function is generalized for both periodic and non-periodic systems (distinguished by atoms.pbc). Especially useful for generating high coverage patterns. All surfaces and nanoparticles are supported (even with surface adsorbates already, such as metal oxides).
- Parameters
atoms (ase.Atoms object) – The nanoparticle or surface slab onto which the adsorbates are added. Accept any ase.Atoms object. No need to be built-in.
adsorbate_species (str or list of strs) – A list of adsorbate species to be randomly added to the surface.
species_probabilities (dict, default None) – A dictionary that contains keys of each adsorbate species and values of their probabilities of adding onto the surface.
adsorption_sites (acat.adsorption_sites.ClusterAdsorptionSites object or acat.adsorption_sites.SlabAdsorptionSites object or the corresponding pickle file path, default None) – Provide the built-in adsorption sites class to accelerate the pattern generation. If this is not provided, the arguments for identifying adsorption sites can still be passed in by **kwargs.
surface (str, default None) – The surface type (crystal structure + Miller indices). If the structure is a periodic surface slab, this is required when adsorption_sites is not provided. If the structure is a nanoparticle, the function only add adsorbates to the sites on the specified surface. For periodic slabs, a user-defined customized surface object can also be used, but note that the identified site types will only include ‘ontop’, ‘bridge’, ‘3fold’ and ‘4fold’.
min_adsorbate_distance (float, default 1.5) – The minimum distance constraint between two atoms that belongs to two adsorbates.
site_types (str or list of strs, default None) – The site type(s) that the adsorbates should be added to. Consider all sites if not specified.
heights (dict, default acat.settings.site_heights) – A dictionary that contains the adsorbate height for each site type. Use the default height settings if the height for a site type is not specified.
site_preference (str or list of strs, defualt None) – The site type(s) that has higher priority to attach adsorbates.
Example 1
To add CO randomly onto a cuboctahedron with a minimum adsorbate distance of 5 Angstrom:
>>> from acat.build.adlayer import min_dist_coverage_pattern as mindcp >>> from ase.cluster import Octahedron >>> from ase.visualize import view >>> atoms = Octahedron('Au', length=9, cutoff=4) >>> atoms.center(vacuum=5.) >>> pattern = mindcp(atoms, adsorbate_species='CO', ... min_adsorbate_distance=5.) >>> view(pattern)Output:

Example 2
To add C, N, O randomly onto a hcp0001 surface slab with probabilities of 0.25, 0.25, 0.5, respectively, and a minimum adsorbate distance of 2 Angstrom:
>>> from acat.build.adlayer import min_dist_coverage_pattern as mindcp >>> from ase.build import hcp0001 >>> from ase.visualize import view >>> atoms = hcp0001('Ru', (8, 8, 4), vacuum=5.) >>> atoms.center(axis=2) >>> pattern = mindcp(atoms, adsorbate_species=['C','N','O'], ... species_probabilities={'C': 0.25, ... 'N': 0.25, ... 'O': 0.5}, ... surface='hcp0001', ... min_adsorbate_distance=2.) >>> view(pattern)Output:

Example 3
To add H, OH and H2O randomly onto a fcc100 Ni2Cu surface slab on both top and bottom interfaces (
bulk waterin between) with probabilities of 0.25, 0.25, 0.5, respectively, and a minimum adsorbate distance of 2 Angstrom:>>> from acat.build.adlayer import min_dist_coverage_pattern as mindcp >>> from ase.io import read >>> from ase.build import fcc100 >>> from ase.visualize import view >>> water_bulk = read('water_bulk.xyz') >>> water_bulk.center(vacuum=11., axis=2) >>> slab = fcc100('Ni', (4, 4, 8), vacuum=9.5, periodic=True) >>> for atom in slab: ... if atom.index % 3 == 0: ... atom.symbol = 'Cu' >>> slab.translate(-slab.cell[2] / 2) >>> slab.wrap() >>> atoms = slab + water_bulk >>> pattern = mindcp(atoms, adsorbate_species=['H','OH','OH2'], ... species_probabilities={'H': 0.25, ... 'OH': 0.25, ... 'OH2': 0.5}, ... surface='fcc100', ... min_adsorbate_distance=2., ... both_sides=True, ... surrogate_metal='Ni') >>> view(pattern)Output:

Generate alloy chemical orderings
The SymmetricClusterOrderingGenerator class
- class acat.build.ordering.SymmetricClusterOrderingGenerator(atoms, elements, symmetry='spherical', cutoff=1.0, secondary_symmetry=None, secondary_cutoff=1.0, composition=None, groups=None, save_groups=False, trajectory='orderings.traj', append_trajectory=False)[source]
SymmetricClusterOrderingGenerator is a class for generating symmetric chemical orderings for a nanoalloy. There is no limitation of the number of metal components. Please always align the z direction to the symmetry axis of the nanocluster.
- Parameters
atoms (ase.Atoms object) – The nanoparticle to use as a template to generate symmetric chemical orderings. Accept any ase.Atoms object. No need to be built-in.
elements (list of strs) – The metal elements of the nanoalloy.
symmetry (str, default 'spherical') –
Support 9 symmetries:
’spherical’: spherical symmetry (groups defined by the distances to the geometric center);
’cylindrical’: cylindrical symmetry around z axis (groups defined by the distances to the z axis);
’planar’: planar symmetry around z axis (groups defined by the z coordinates), common for phase-separated nanoalloys;
’mirror_planar’: mirror planar symmetry around both z and xy plane (groups defined by the absolute z coordinate), high symmetry subset of ‘planar’; ‘circular’ = ring symmetry around z axis (groups defined by both z coordinate and distance to z axis);
’mirror_circular’: mirror ring symmetry around both z and xy plane (groups defined by both absolute z coordinate and distance to z axis);
’chemical’: symmetry w.r.t chemical environment (groups defined by the atomic energies given by a Gupta potential)
’geometrical’: symmetry w.r.t geometrical environment (groups defined by vertex / edge / fcc111 / fcc100 / bulk identified by CNA analysis);
’concentric’: conventional definition of the concentric shells (surface / subsurface, subsubsurface, …, core).
cutoff (float, default 1.0) – Maximum thickness (in Angstrom) of a single group. The thickness is calculated as the difference between the “distances” of the closest-to-center atoms in two neighbor groups. Note that the criterion of “distance” depends on the symmetry. This parameter works differently if the symmetry is ‘chemical’, ‘geometrical’ or ‘concentric’. For ‘chemical’ it is defined as the maximum atomic energy difference (in eV) of a single group predicted by a Gupta potential. For ‘geometrical’ and ‘concentric’ it is defined as the cutoff radius (in Angstrom) for CNA, and a reasonable cutoff based on the lattice constant of the material will automatically be used if cutoff <= 1. Use a larger cutoff if the structure is distorted.
secondary_symmetry (str, default None) – Add a secondary symmetry check to define groups hierarchically. For example, even if two atoms are classifed in the same group defined by the primary symmetry, they can still end up in different groups if they fall into two different groups defined by the secondary symmetry. Support 7 symmetries: ‘spherical’, ‘cylindrical’, ‘planar’, ‘mirror_planar’, ‘chemical’, ‘geometrical’ and ‘concentric’. Note that secondary symmetry has the same importance as the primary symmetry, so you can set either of the two symmetries of interest as the secondary symmetry. Useful for combining symmetries of different types (e.g. circular + chemical) or combining symmetries with different cutoffs.
secondary_cutoff (float, default 1.0) – Same as cutoff, except that it is for the secondary symmetry.
composition (dict, default None) – Generate symmetric orderings only at a certain composition. The dictionary contains the metal elements as keys and their concentrations as values. Generate orderings at all compositions if not specified.
groups (list of lists, default None) – Provide the user defined symmetry equivalent groups as a list of lists of atom indices. Useful for generating structures with symmetries that are not supported.
save_groups (bool, default False) – Whether to save the site groups in atoms.info[‘data’][‘groups’] for each generated structure.
trajectory (str, default 'orderings.traj') – The name of the output ase trajectory file.
append_trajectory (bool, default False) – Whether to append structures to the existing trajectory.
- get_sorted_indices(symmetry)[source]
Returns the indices sorted by the metric that defines different groups, together with the corresponding vlues, given a specific symmetry. Returns the indices sorted by geometrical environment if symmetry=’geometrical’. Returns the indices sorted by surface, subsurface, subsubsurface, …, core if symmetry=’concentric’.
- Parameters
symmetry (str) – Support 7 symmetries: spherical, cylindrical, planar, mirror_planar, chemical, geometrical, concentric.
- get_groups()[source]
Get the groups (a list of lists of atom indices) of all symmetry-equivalent atoms.
- run(max_gen=None, mode='stochastic', eps=0.01, unique=False, hmax=2, verbose=False)[source]
Run the chemical ordering generator.
- Parameters
max_gen (int, default None) – Maximum number of chemical orderings to generate. Running forever (until exhaustive for systematic search) if not specified.
mode (str, default 'stochastic') –
‘systematic’: enumerate all possible unique chemical orderings. Recommended when there are not many groups. Switch to stochastic mode automatically if the number of groups is more than 20 (30 if composition is fixed since there are much fewer structures).
’stochastic’: sample chemical orderings stochastically. Duplicate structures can be generated. Recommended when there are many groups. A greedy algorithm is employed if the composition is fixed, which might result in slightly different compositions. This can be controled by eps.
eps (float, default 0.01) – The tolerance of the concentration for each element (the target concentration range is [c-eps, c+eps]). Only relevant for generating fixed-composition symmetric nanoalloys using ‘stochastic’ mode. Set to a small value, e.g. 0.005, if you want the exact composition accurate to one atom. Please use a larger eps if the concentrations you specified in compositions are not accurate enough.
unique (bool, default False) – Whether to discard duplicate patterns based on graph automorphism. The Weisfeiler-Lehman subtree kernel is used to check identity.
hmax (int, default 2) – Maximum number of iterations for color refinement. Only relevant if unique=True.
verbose (bool, default False) – Whether to print out information about number of groups and number of generated structures.
Example 1
To generate 100 symmetric chemical orderings for truncated octahedral NiPt nanoalloys with spherical symmetry:
>>> from acat.build.ordering import SymmetricClusterOrderingGenerator as SCOG >>> from ase.cluster import Octahedron >>> from ase.io import read >>> from ase.visualize import view >>> atoms = Octahedron('Ni', length=8, cutoff=3) >>> scog = SCOG(atoms, elements=['Ni', 'Pt'], symmetry='spherical') >>> scog.run(max_gen=100, verbose=True) >>> images = read('orderings.traj', index=':') >>> view(images)Output:
10 symmetry-equivalent groups classified100 symmetric chemical orderings generated
Example 2
To systematically generate 50 symmetric chemical orderings for quaternary truncated octahedral Ni0.4Cu0.3Pt0.2Au0.1 nanoalloys with mirror circular symmetry:
>>> from acat.build.ordering import SymmetricClusterOrderingGenerator as SCOG >>> from ase.cluster import Octahedron >>> from ase.io import read >>> from ase.visualize import view >>> atoms = Octahedron('Ni', 7, 2) >>> scog = SCOG(atoms, elements=['Ni', 'Cu', 'Pt', 'Au'], ... symmetry='mirror_circular', ... composition={'Ni': 0.4, 'Cu': 0.3, 'Pt': 0.2, 'Au': 0.1}) >>> scog.run(max_gen=50, mode='systematic', verbose=True) >>> images = read('orderings.traj', index=':') >>> view(images)Output:
25 symmetry-equivalent groups classified50 symmetric chemical orderings generated
Example 3
To stochastically generate 50 symmetric chemical orderings for quaternary truncated octahedral Ni0.4Cu0.3Pt0.2Au0.1 nanoalloys with mirror circular symmetry:
>>> from acat.build.ordering import SymmetricClusterOrderingGenerator as SCOG >>> from ase.cluster import Octahedron >>> from ase.io import read >>> from ase.visualize import view >>> atoms = Octahedron('Ni', 7, 2) >>> scog = SCOG(atoms, elements=['Ni', 'Cu', 'Pt', 'Au'], ... symmetry='mirror_circular', ... composition={'Ni': 0.4, 'Cu': 0.3, 'Pt': 0.2, 'Au': 0.1}) >>> scog.run(max_gen=50, mode='stochastic', verbose=True) >>> images = read('orderings.traj', index=':') >>> view(images)Output:
25 symmetry-equivalent groups classified50 symmetric chemical orderings generated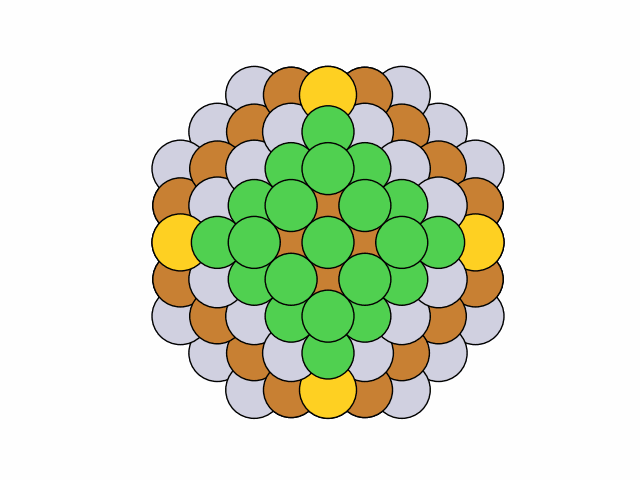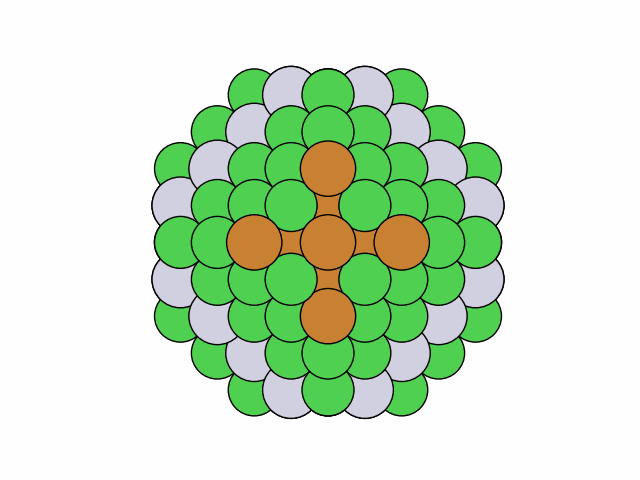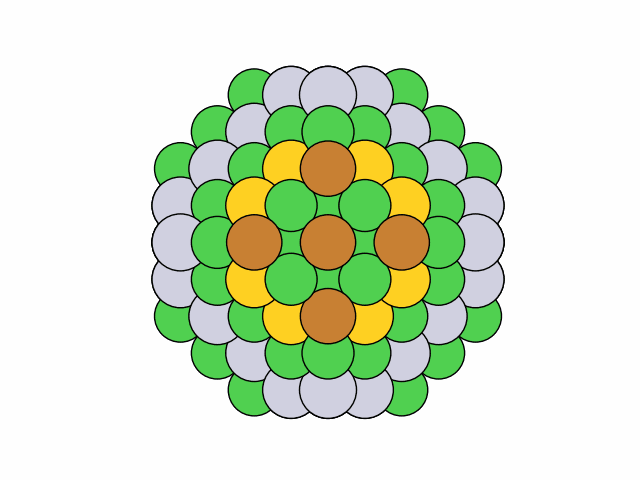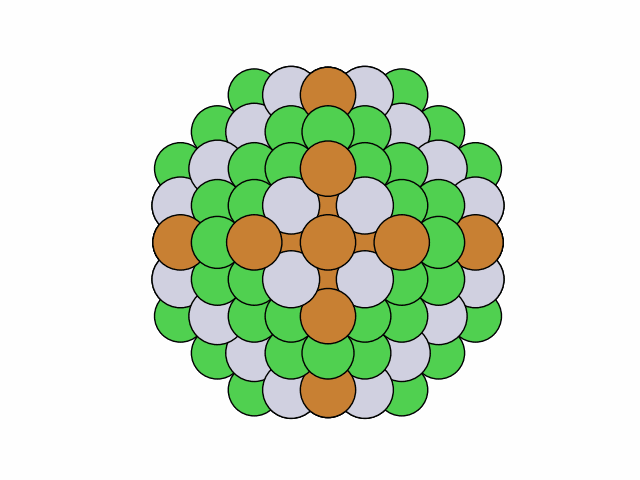
Example 4
Sometimes it is also useful to get the structure of each group. For instance, to visualize the concentric shells of a truncated octahedral nanoparticle:
>>> from acat.build.ordering import SymmetricClusterOrderingGenerator as SCOG >>> from ase.cluster import Octahedron >>> from ase.visualize import view >>> atoms = Octahedron('Ni', 12, 5) >>> scog = SCOG(atoms, elements=['Ni', 'Pt'], symmetry='concentric') >>> groups = scog.get_groups() >>> images = [atoms[g] for g in groups] >>> view(images)Output:

The SymmetricSlabOrderingGenerator class
- class acat.build.ordering.SymmetricSlabOrderingGenerator(atoms, elements, symmetry='translational', repeating_size=(1, 1), bisect_vector=None, composition=None, groups=None, save_groups=False, dtol=0.01, ztol=0.1, trajectory='orderings.traj', append_trajectory=False)[source]
SymmetricSlabOrderingGenerator is a class for generating symmetric chemical orderings for a alloy surface slab. There is no limitation of the number of metal components.
- Parameters
atoms (ase.Atoms object) – The surface slab to use as a template to generate symmetric chemical orderings. Accept any ase.Atoms object. No need to be built-in.
elements (list of strs) – The metal elements of the alloy catalyst.
symmetry (str, default 'translational') –
Support 4 symmetries:
’translational’: translational symmetry with symmetry equivalent groups depending on repeating_size.
’inversion’: centrosymmetry with symmetry equivalent groups depending on the distance to the geometric center. Only works for certain number of slab layers (e.g. 5) and orthogonal cell.
’vertical_mirror’: vertical mirror plane symmetry with symmetry equivalent groups depending on the vertical plane in which the bisect_vector lies.
’horizontal_mirror’: horizontal mirror plane symmetry with symmetry equivalent groups depending on the xy plane bisecting the slab. Only works for certain number of slab layers (e.g. 5) and orthogonal cell.
bisect_vector (numpy.array or list, default None) – The 2d (or 3d) vector that lies in the vertical mirror plane. Only relevant for vertical_mirror symmetry.
repeating_size (list of ints or tuple of ints, default (1, 1)) – The multiples that describe the size of the repeating pattern on the surface. Symmetry-equivalent atoms are grouped by the multiples in the x and y directions. The x or y length of the cell must be this multiple of the distance between each pair of symmetry-equivalent atoms. Larger reducing size generates fewer structures. For translational symmetry please change repeating_size to larger numbers.
composition (dict, default None) – Generate symmetric orderings only at a certain composition. The dictionary contains the metal elements as keys and their concentrations as values. Generate orderings at all compositions if not specified.
groups (list of lists, default None) – Provide the user defined symmetry equivalent groups as a list of lists of atom indices. Useful for generating structures with symmetries that are not supported.
save_groups (bool, default False) – Whether to save the site groups in atoms.info[‘data’][‘groups’] for each generated structure.
dtol (float, default 0.01) – The distance tolerance (in Angstrom) when comparing distances. Use a larger value if the structure is distorted.
ztol (float, default 0.1) – The tolerance (in Angstrom) when comparing z values. Use a larger ztol if the structure is distorted.
trajectory (str, default 'orderings.traj') – The name of the output ase trajectory file.
append_trajectory (bool, default False) – Whether to append structures to the existing trajectory.
- get_groups()[source]
Get the groups (a list of lists of atom indices) of all symmetry-equivalent atoms.
- run(max_gen=None, mode='stochastic', eps=0.01, unique=False, hmax=2, verbose=False)[source]
Run the chemical ordering generator.
- Parameters
max_gen (int, default None) – Maximum number of chemical orderings to generate. Running forever (until exhaustive for systematic search) if not specified.
mode (str, default 'stochastic') –
‘systematic’: enumerate all possible unique chemical orderings. Recommended when there are not many groups. Switch to stochastic mode automatically if the number of groups is more than 20 (30 if composition is fixed since there are much fewer structures).
’stochastic’: sample chemical orderings stochastically. Duplicate structures can be generated. Recommended when there are many groups. A greedy algorithm is employed if the composition is fixed, which might result in slightly different compositions. This can be controled by eps.
eps (float, default 0.01) – The tolerance of the concentration for each element (the target concentration range is [c-eps, c+eps]). Only relevant for generating fixed-composition symmetric nanoalloys using ‘stochastic’ mode. Set to a small value, e.g. 0.005, if you want the exact composition accurate to one atom. Please use a larger eps if the concentrations you specified in compositions are not accurate enough.
unique (bool, default False) – Whether to discard duplicate patterns based on graph automorphism. The Weisfeiler-Lehman subtree kernel is used to check identity.
hmax (int, default 2) – Maximum number of iterations for color refinement. Only relevant if unique=True.
verbose (bool, default False) – Whether to print out information about number of groups and number of generated structures.
Example 1
To stochastically generate 20 chemical orderings with vertical mirror plane symmetry w.r.t. the bisect vector [11.200, 6.467] for binary NixPt1-x fcc111 surface slabs without duplicates:
>>> from acat.build.ordering import SymmetricSlabOrderingGenerator as SSOG >>> from ase.build import fcc111 >>> from ase.io import read >>> from ase.visualize import view >>> import numpy as np >>> atoms = fcc111('Ni', (4, 4, 5), vacuum=5.) >>> atoms.center(axis=2) >>> ssog = SSOG(atoms, elements=['Ni', 'Pt'], ... symmetry='vertical_mirror', ... bisect_vector=np.array([11.200, 6.467])) >>> ssog.run(max_gen=20, mode='stochastic', unique=True, verbose=True) >>> images = read('orderings.traj', index=':') >>> view(images)Output:
50 symmetry-equivalent groups classified20 symmetric chemical orderings generated
Example 2
To stochastically generate 50 chemical orderings with translational symmetry for ternary NixPtyAu1-x-y fcc111 surface slabs:
>>> from acat.build.ordering import SymmetricSlabOrderingGenerator as SSOG >>> from ase.build import fcc111 >>> from ase.io import read >>> from ase.visualize import view >>> atoms = fcc111('Ni', (6, 6, 4), vacuum=5.) >>> atoms.center(axis=2) >>> ssog = SSOG(atoms, elements=['Ni', 'Pt', 'Au'], ... symmetry='translational', ... repeating_size=(3, 3)) >>> ssog.run(max_gen=50, mode='stochastic', verbose=True) >>> images = read('orderings.traj', index=':') >>> view(images)Output:
16 symmetry-equivalent groups classified50 symmetric chemical orderings generated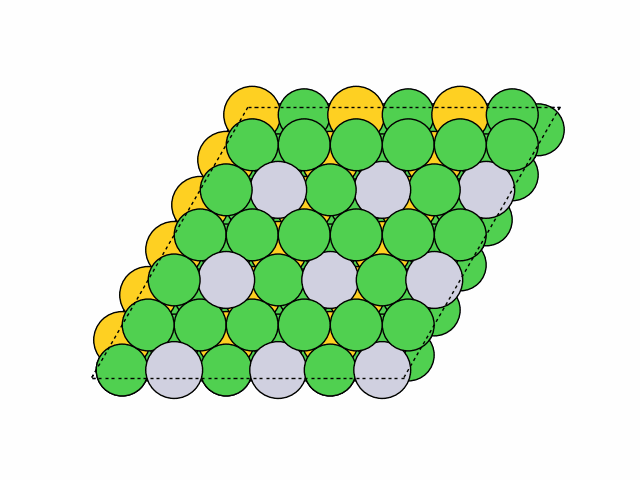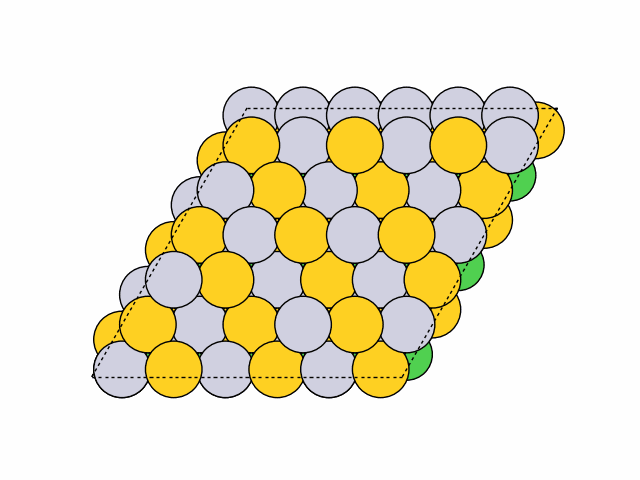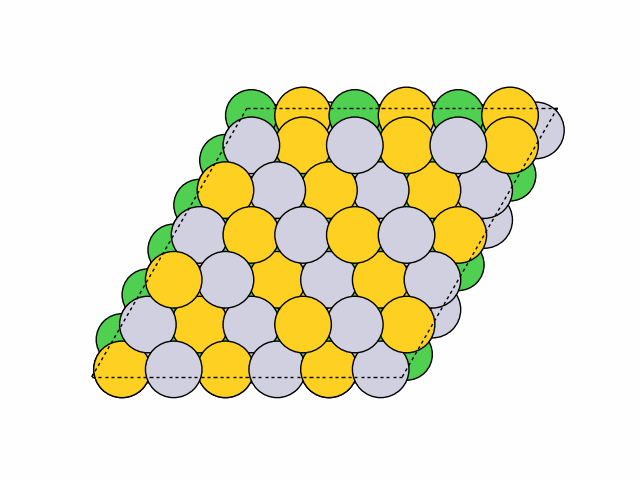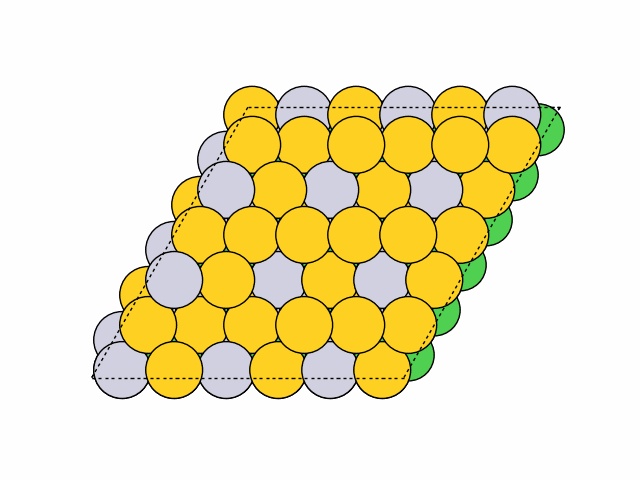
Example 3
To systematically generate 50 chemical orderings with translational symmetry for Ni0.75Pt0.25 fcc110 surface slabs:
>>> from acat.build.ordering import SymmetricSlabOrderingGenerator as SSOG >>> from ase.build import fcc110 >>> from ase.io import read >>> from ase.visualize import view >>> atoms = fcc110('Ni', (4, 4, 4), vacuum=5.) >>> atoms.center(axis=2) >>> ssog = SSOG(atoms, elements=['Ni', 'Pt'], ... symmetry='translational', ... composition={'Ni': 0.75, 'Pt': 0.25}, ... repeating_size=(2, 2)) >>> ssog.run(max_gen=50, mode='systematic', verbose=True) >>> images = read('orderings.traj', index=':') >>> view(images)Output:
16 symmetry-equivalent groups classified50 symmetric chemical orderings generated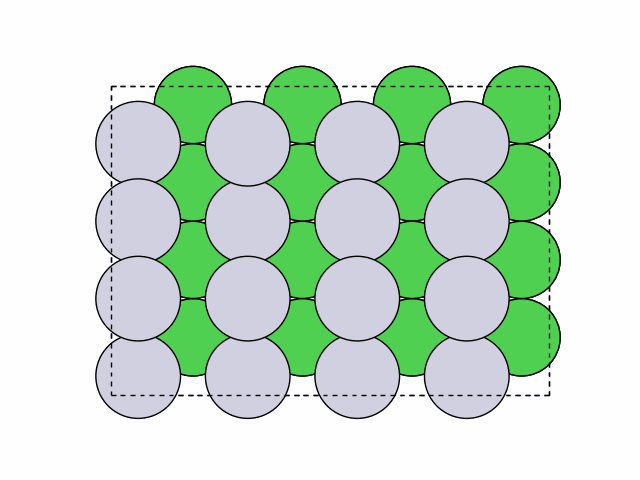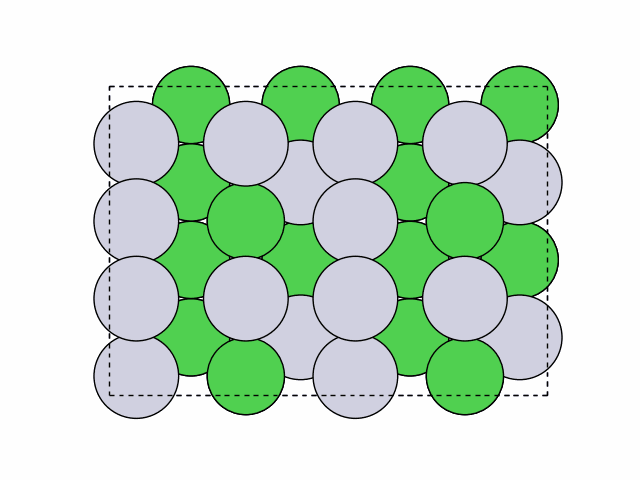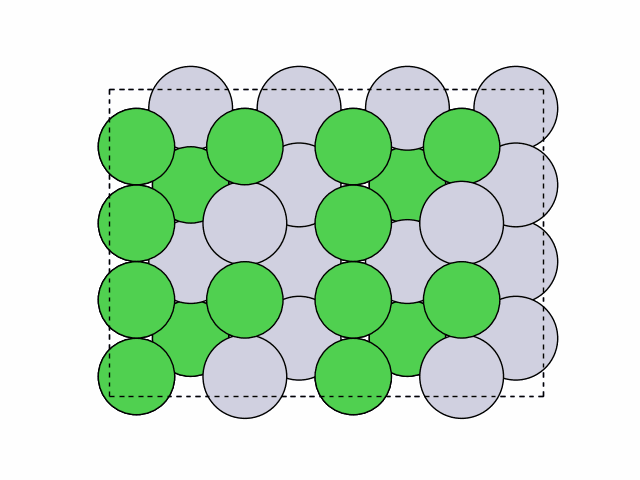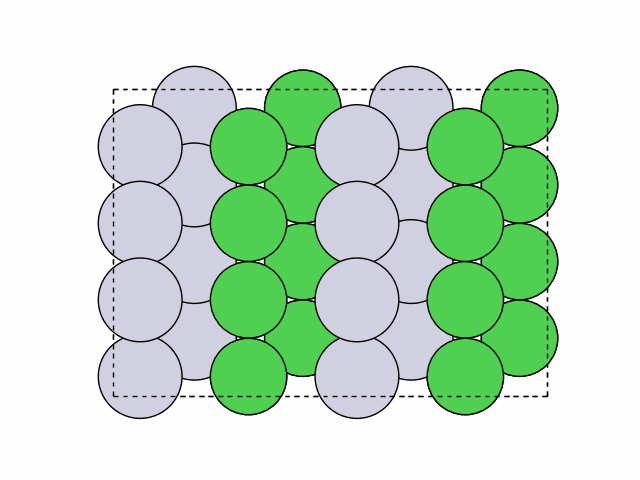
Example 4
To stochastically generate 50 chemical orderings with translational symmetry for Ni0.75Pt0.25 fcc110 surface slabs:
>>> from acat.build.ordering import SymmetricSlabOrderingGenerator as SSOG >>> from ase.build import fcc110 >>> from ase.io import read >>> from ase.visualize import view >>> atoms = fcc110('Ni', (4, 4, 4), vacuum=5.) >>> atoms.center(axis=2) >>> ssog = SSOG(atoms, elements=['Ni', 'Pt'], ... symmetry='translational', ... composition={'Ni': 0.75, 'Pt': 0.25}, ... repeating_size=(2, 2)) >>> ssog.run(max_gen=50, mode='stochastic', verbose=True) >>> images = read('orderings.traj', index=':') >>> view(images)Output:
16 symmetry-equivalent groups classified50 symmetric chemical orderings generated
The RandomOrderingGenerator class
- class acat.build.ordering.RandomOrderingGenerator(atoms, elements, composition=None, trajectory='orderings.traj', append_trajectory=False)[source]
RandomOrderingGenerator is a class for generating random chemical orderings for an alloy catalyst (can be either bulk, slab or nanoparticle). The function is generalized for both periodic and non-periodic systems, and there is no limitation of the number of metal components.
- Parameters
atoms (ase.Atoms object) – The nanoparticle or surface slab to use as a template to generate random chemical orderings. Accept any ase.Atoms object. No need to be built-in.
elements (list of strs) – The metal elements of the alloy catalyst.
composition (dict, default None) – Generate random orderings only at a certain composition. The dictionary contains the metal elements as keys and their concentrations as values. Generate orderings at all compositions if not specified.
trajectory (str, default 'orderings.traj') – The name of the output ase trajectory file.
append_trajectory (bool, default False) – Whether to append structures to the existing trajectory.
- randint_with_sum()[source]
Return a randomly chosen list of N positive integers i summing to the number of atoms. N is the number of elements. Each such list is equally likely to occur.
- run(num_gen, unique=False, hmax=2)[source]
Run the chemical ordering generator.
- Parameters
num_gen (int) – Number of chemical orderings to generate.
unique (bool, default False) – Whether to discard duplicate patterns based on graph automorphism. The Weisfeiler-Lehman subtree kernel is used to check identity.
hmax (int, default 2) – Maximum number of iterations for color refinement. Only relevant if unique=True.
Example 1
To generate 50 random chemical orderings for icosahedral Ni0.5Pt0.2Au0.3 nanoalloys:
>>> from acat.build.ordering import RandomOrderingGenerator as ROG >>> from ase.cluster import Icosahedron >>> from ase.io import read >>> from ase.visualize import view >>> atoms = Icosahedron('Ni', noshells=4) >>> rog = ROG(atoms, elements=['Ni', 'Pt', 'Au'], ... composition={'Ni': 0.5, 'Pt': 0.2, 'Au': 0.3}) >>> rog.run(num_gen=50) >>> images = read('orderings.traj', index=':') >>> view(images)Output:

Example 2
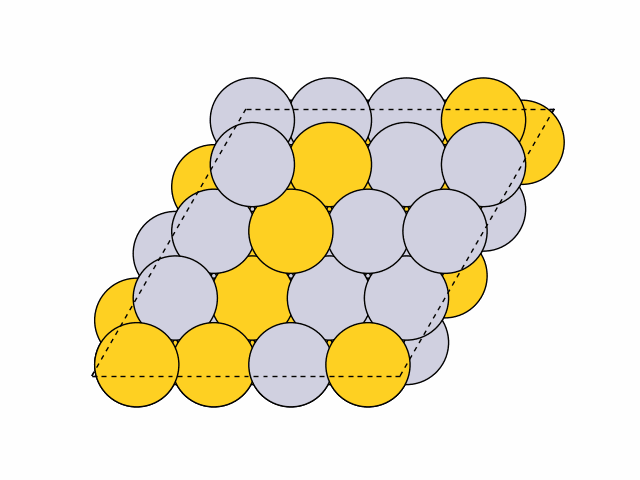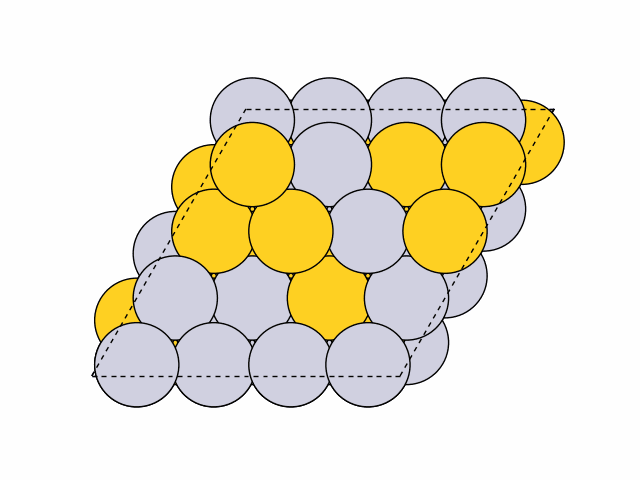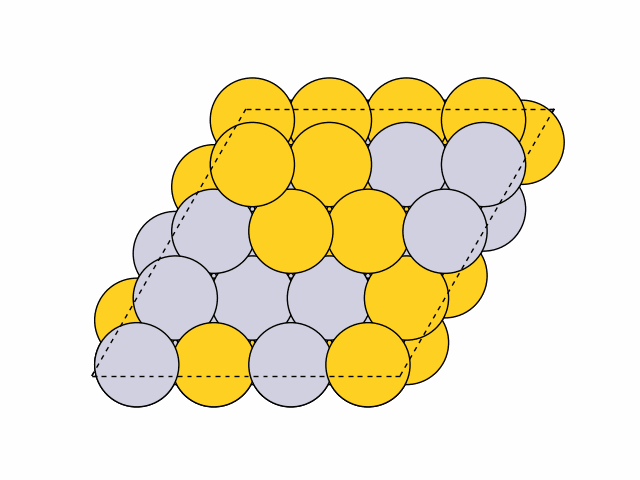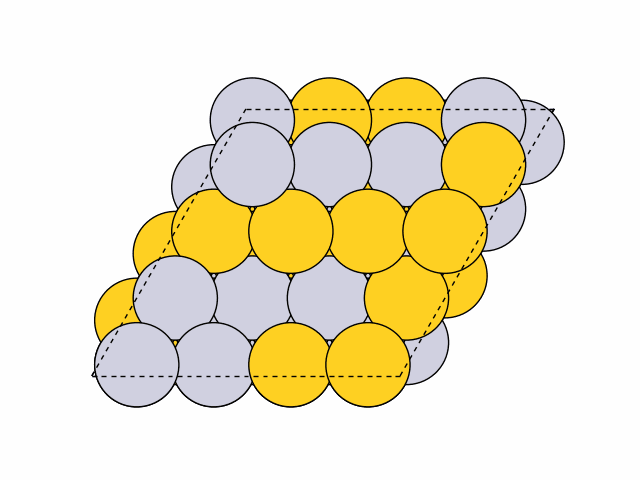
To generate 50 random chemical orderings for Pt0.5Au0.5 fcc111 surface slabs without duplicates:
>>> from acat.build.ordering import RandomOrderingGenerator as ROG >>> from ase.build import fcc111 >>> from ase.io import read >>> from ase.visualize import view >>> atoms = fcc111('Ni', (4, 4, 4), vacuum=5.) >>> atoms.center(axis=2) >>> rog = ROG(atoms, elements=['Pt', 'Au'], ... composition={'Pt': 0.5, 'Au': 0.5}) >>> rog.run(num_gen=50, unique=True) >>> images = read('orderings.traj', index=':') >>> view(images)Output:
Example 3
Although the code is written for metal alloys, it can also be used to generate random mixed metal oxides with some simple work around. Below is an example of generating 50 random equiatomic anatase Ti0.2Pt0.2Pd0.2Ag0.2Au0.2O2(101) high-entropy oxide surfaces:
>>> from acat.build.ordering import RandomOrderingGenerator as ROG >>> from ase.spacegroup import crystal >>> from ase.build import surface >>> from ase.io import read >>> from ase.visualize import view >>> a, c = 3.862, 9.551 >>> TiO2 = crystal(['Ti', 'O'], basis=[(0 ,0, 0), (0, 0, 0.208)], ... spacegroup=141, cellpar=[a, a, c, 90, 90, 90]) >>> atoms = surface(TiO2, (1, 0, 1), 4, vacuum=5) >>> atoms *= (1,2,1) >>> atoms.center(axis=2) >>> # Distinguish metal and oxygen atoms >>> metal_ids, oxygen_ids = [], [] >>> for i, atom in enumerate(atoms): ... if atom.symbol == 'O': ... oxygen_ids.append(i) ... else: ... metal_ids.append(i) >>> metal_atoms, oxygen_atoms = atoms[metal_ids], atoms[oxygen_ids] >>> # Prepare sorted_ids for sorting atomic indices back to original order >>> sorted_ids = [0] * len(atoms) >>> for k, v in enumerate(metal_ids + oxygen_ids): ... sorted_ids[v] = k >>> rog = ROG(metal_atoms, elements=['Ti','Pt','Pd','Ag','Au'], ... composition={'Ti':0.2,'Pt':0.2,'Pd':0.2,'Ag':0.2,'Au':0.2}) >>> rog.run(num_gen=50) >>> metal_images = read('orderings.traj', index=':') >>> images = [(a + oxygen_atoms)[sorted_ids] for a in metal_images] >>> view(images)Output:
